From DNA, RNA and mRNA to tRNAs, guide RNAs, aptamers, duplexes and specialised nucleic acid complexes, Vici.bio helps teams predict structure, model co-folding, optimise sequences and analyse deeper nucleic-acid interactions.

Predict nucleic acid structures, model duplexes, hairpins and larger assemblies, and support harder cases like protein-DNA, protein-RNA, ligand-bound RNA and broader co-folding workflows.
Analyse mRNA, tRNAs, guide RNAs, aptamers, ribozymes and other structured RNA systems where local motifs, loops, pairing patterns and binding context all matter.
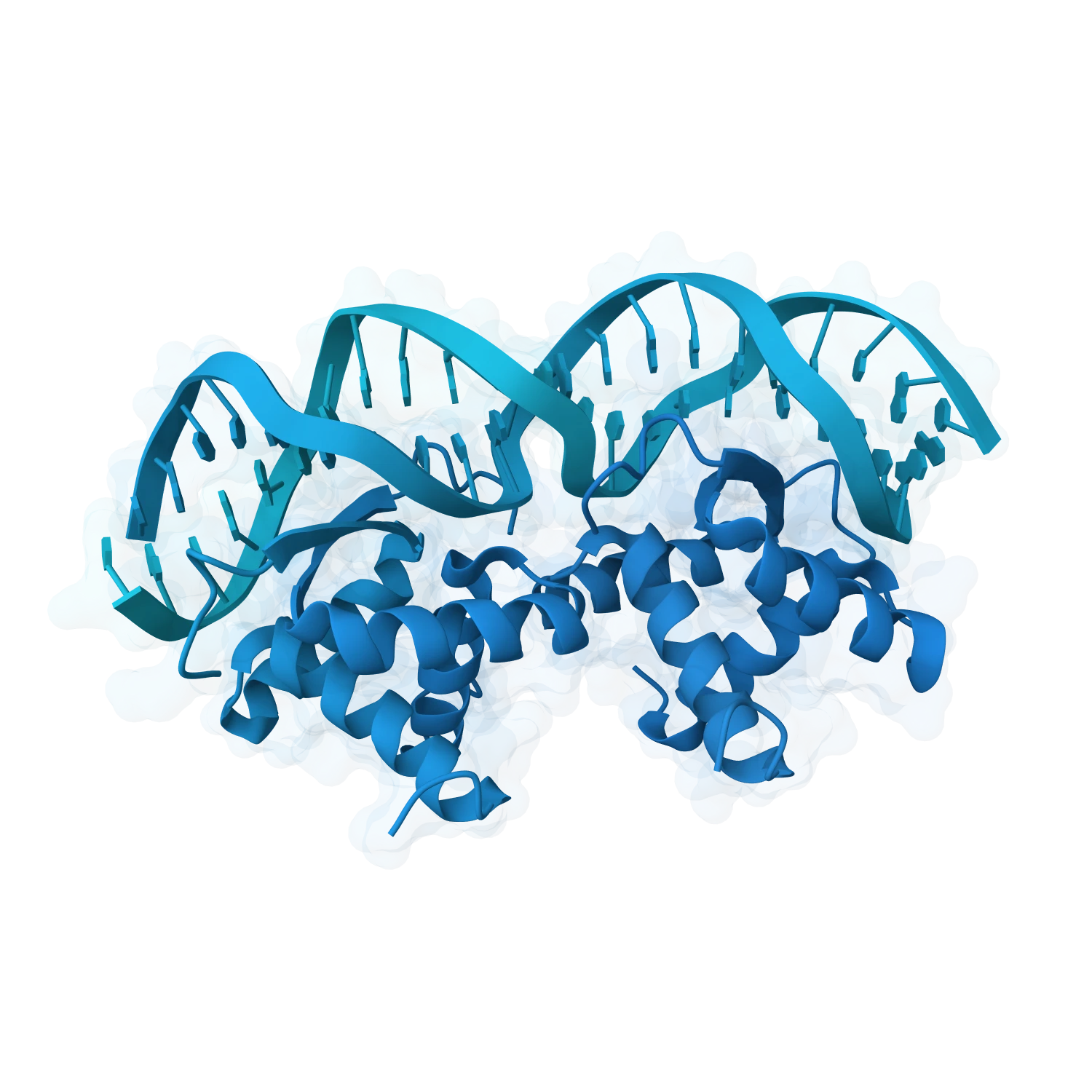
Explore protein-DNA and protein-RNA interfaces, inspect binding contacts, compare structural states and support workflows where recognition, specificity and nucleic-acid conformation must be analysed together.
Use the editor to mutate bases, redesign motifs, optimise oligos, adjust loops, tune guide sequences and move faster from sequence editing to structure-aware analysis.
Support metal ions, small-molecule binders, modified nucleotides, base analogues and other chemically enriched cases where folding, recognition and stability depend on more than sequence alone.

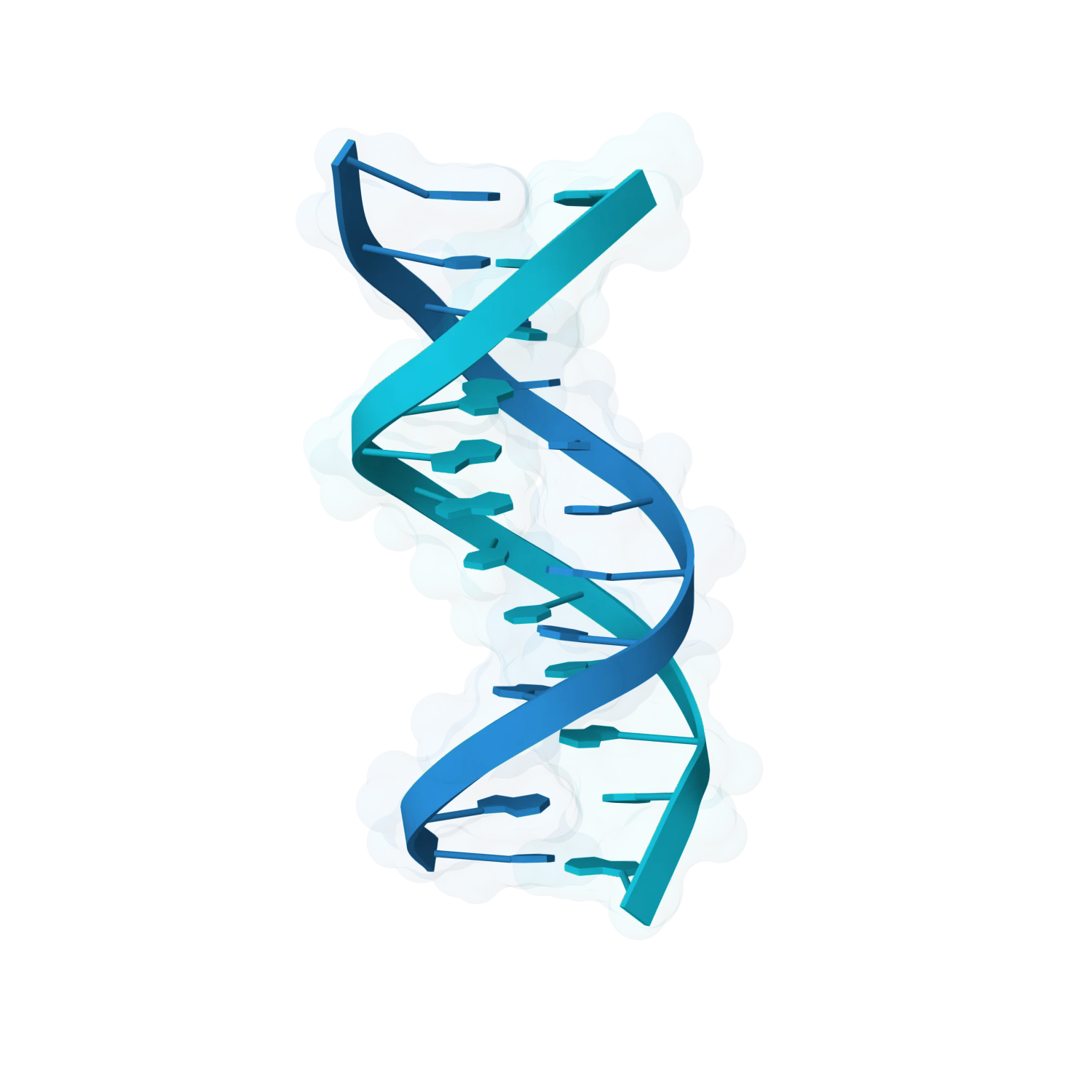
Go beyond the obvious with stem loops, structured motifs, duplex variants, specialised RNA folds and harder nucleic-acid architectures that still need strong modelling, optimisation and interaction support.
Run molecular dynamics, inspect conformational changes, compare flexible states and support nucleic-acid systems that cannot be understood from a single static structure, especially in larger or more dynamic assemblies.
From folding and co-folding to sequence editing, interaction analysis, modified-base modelling and molecular dynamics, build nucleic-acid workflows designed for modern computational biology.